小罗碎碎念
这篇文章提出了用于胎盘组织学全切片图像分析的深度学习框架HAPPY,其核心是通过三阶段流水线实现从细胞到组织结构的层次化建模。

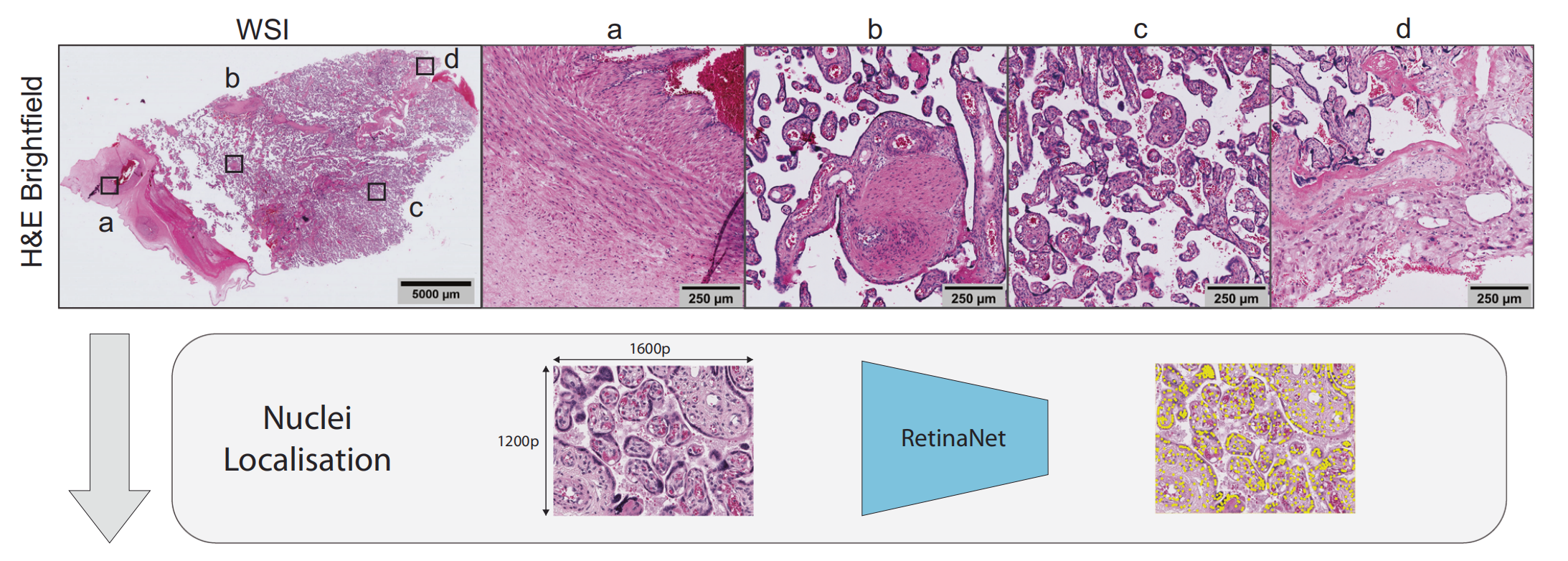
- 首先利用RetinaNet定位细胞核。
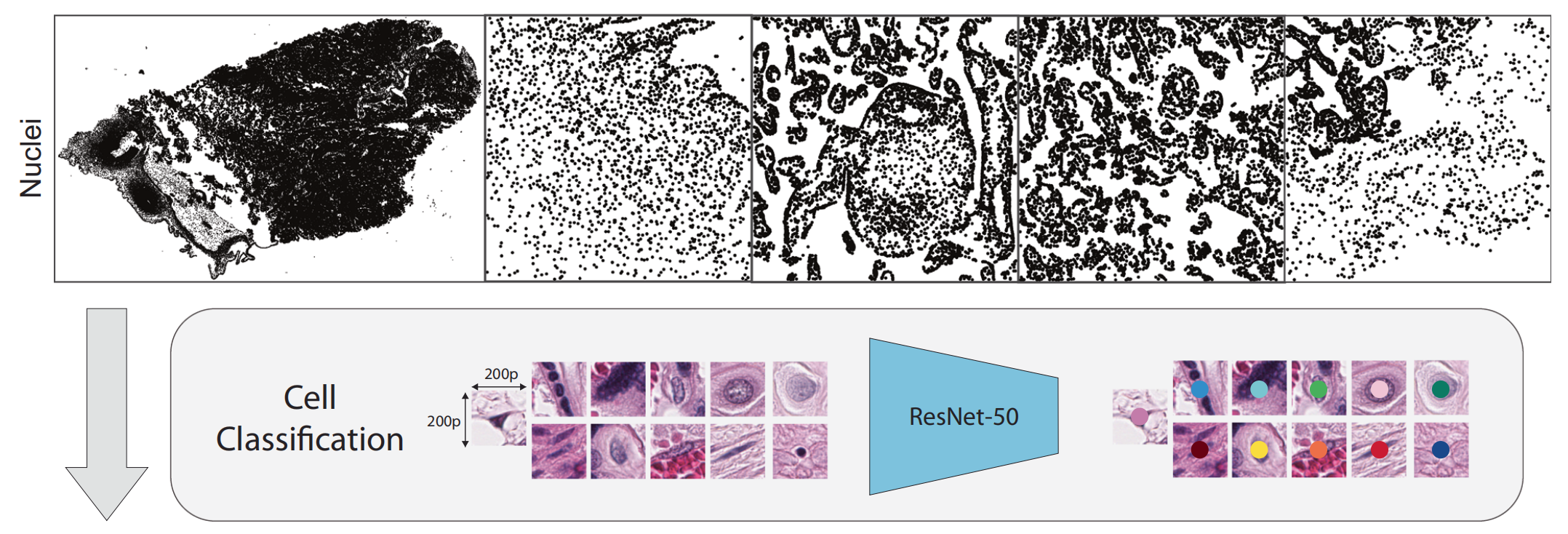
- 再通过ResNet-50分类11种细胞类型
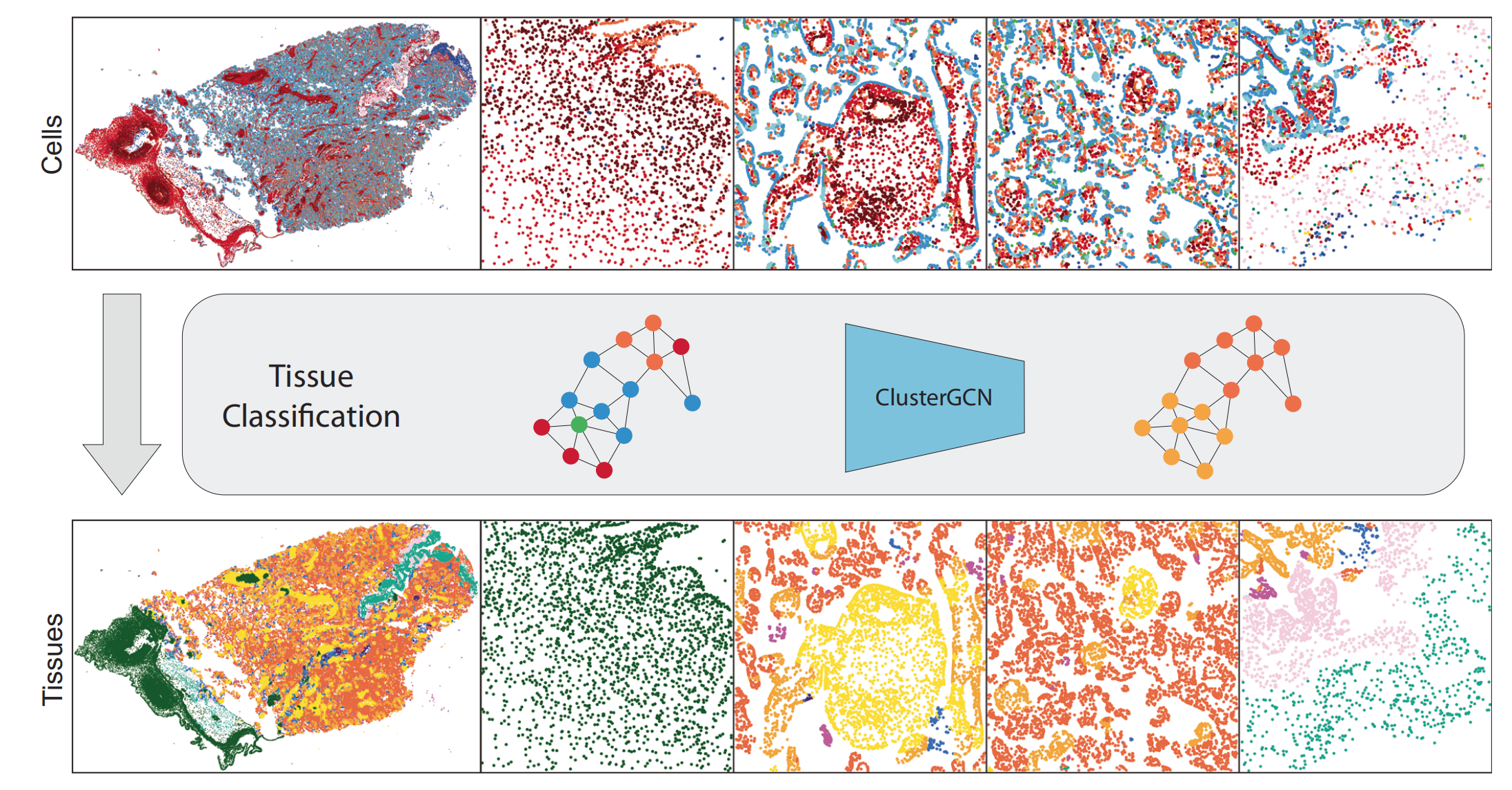
最后基于ClusterGCN图神经网络将细胞聚合成9类显微组织结构,构建全切片空间细胞图。
模型在细胞核检测(F1=0.884)、细胞分类(准确率84.29%)和组织分类(准确率68.34%)上展现出与病理专家相当的性能,且通过动态图区域采样和细胞社区聚合,突破了传统基于图像块方法的分辨率限制,为胎盘组织的单细胞分辨率分析提供了新范式。
交流群
欢迎大家加入【医学AI】交流群,本群设立的初衷是提供交流平台,方便大家后续课题合作。
目前小罗全平台关注量67,000+,交流群总成员1500+,大部分来自国内外顶尖院校/医院,期待您的加入!!
由于近期入群推销人员较多,已开启入群验证,扫码添加我的联系方式,备注姓名-单位-科室/专业,即可邀您入群。
知识星球
对推文中的内容感兴趣,想深入探讨?在处理项目时遇到了问题,无人商量?加入小罗的知识星球,寻找科研道路上的伙伴吧!
一、文献概述
胎盘病理评估对母婴健康管理至关重要,但其异质性和时间变异性给组织学分析带来挑战。本文介绍了一种名为HAPPY(Histology Analysis Pipeline.PY)的深度学习分层方法,用于量化胎盘组织学全切片图像中细胞和显微解剖组织结构的变异性。
| 类别 | 姓名 | 单位 |
|---|---|---|
| 第一作者 | Claudia Vanea | 牛津大学妇女与生殖健康系、牛津大学李嘉诚健康信息与发现中心大数据研究所 |
| 通讯作者 | Claudia Vanea | 牛津大学妇女与生殖健康系、牛津大学李嘉诚健康信息与发现中心大数据研究所 |
| 通讯作者 | Christoffer Nellåker | 牛津大学妇女与生殖健康系、牛津大学李嘉诚健康信息与发现中心大数据研究所 |
1-1:方法与模型架构
HAPPY通过三个阶段实现对胎盘组织的分析:
- 细胞核定位:使用RetinaNet目标检测模型定位全切片图像中的细胞核,在测试集上达到0.884的F1分数,性能与其他器官的先进模型相当。
- 细胞分类:基于ResNet-50的图像分类模型将细胞分为11种类型,总体准确率为84.29%,top-2准确率达94.90%,展现了较高的细胞类型识别能力。
- 组织分类:通过图神经网络(GNN)ClusterGCN构建细胞空间图,将细胞聚合成9种显微组织结构,整体准确率为68.34%,top-2和top-3准确率分别为91.14%和97.10%,能够有效捕捉组织结构的层次关系。
1-2:性能验证与临床应用
- 与病理专家对比:模型在组织分类上的表现与四位围产期病理专家的标注结果具有较强一致性,Cohen’s kappa值为0.61,且模型的PR-AUC值与专家间的kappa值呈显著正相关(R²=0.821),表明其可靠性可与人类专家媲美。
- 健康胎盘基线指标:通过分析30例健康足月胎盘的全切片图像,建立了细胞和组织结构的定量基线,如合胞体滋养层细胞占比>40%,终末绒毛占比30-60%等,为胎盘健康评估提供了新的量化标准。
- 胎盘梗死案例研究:在12例临床显著胎盘梗死的样本中,模型检测到合胞体滋养层细胞、成纤维细胞和血管内皮细胞显著减少,而绒毛外滋养层细胞和白细胞增多,同时纤维蛋白和无血管绒毛比例增加,验证了其在病理检测中的应用价值。
1-3:泛化能力与数据共享
HAPPY通过数据增强(如H&E染色增强)提升了跨机构数据的泛化能力,在不同染色条件下表现稳定。
模型代码、训练数据和预训练模型已开源(https://github.com/Nellaker-group/happy),并提供多器官适用性验证,为胎盘研究和数字病理领域提供了重要工具。
1-4:结论
HAPPY通过深度学习实现了胎盘组织学的高通量、定量分析,突破了传统手动评估的局限性,为胎盘健康和病理机制研究提供了新范式,有望推动围产期医学和生殖健康领域的发展。
二、方法
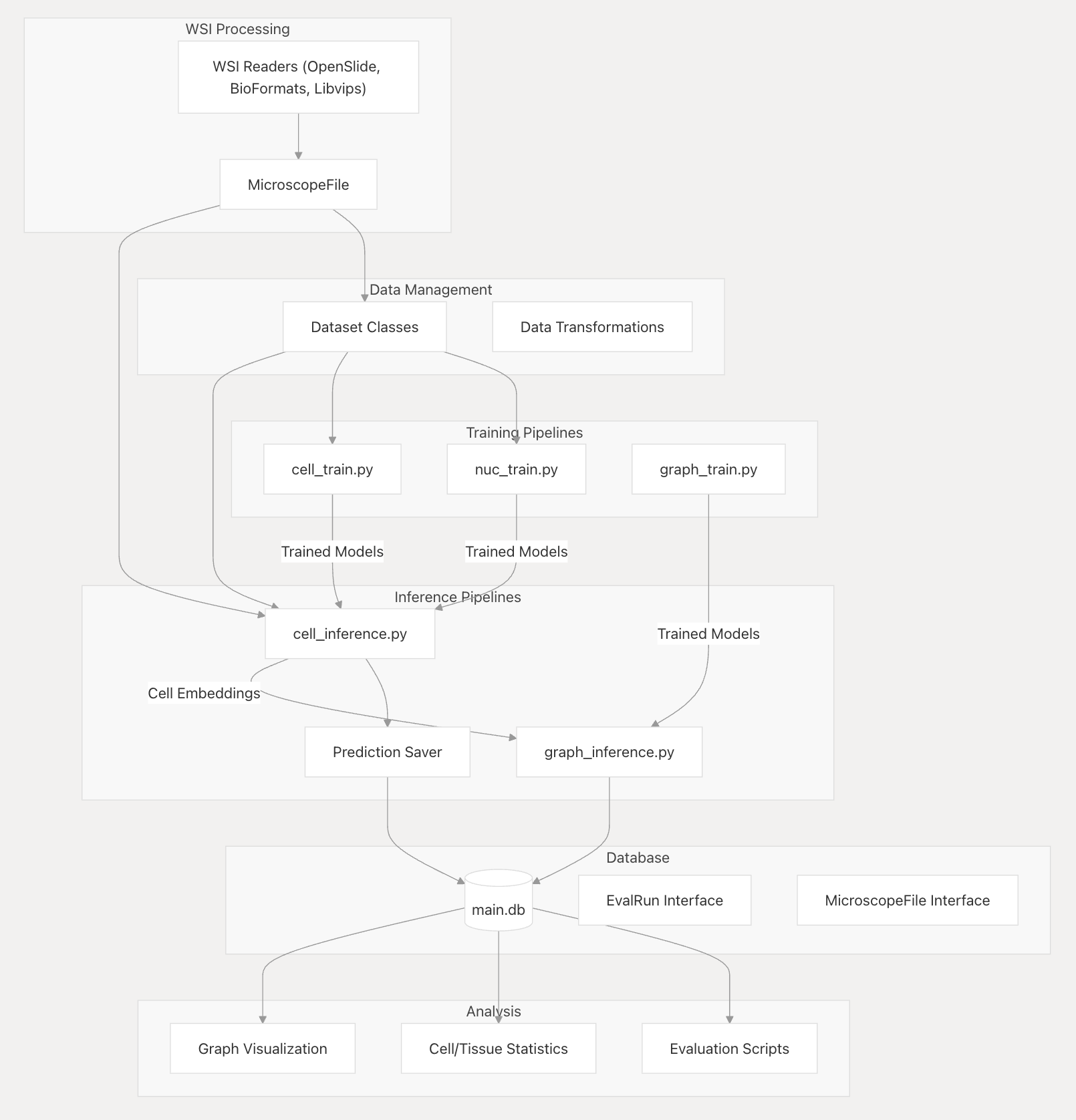
HAPPY作为一个有监督的深度学习流水线,主要分为三个阶段:(i)细胞核定位,(ii)细胞分类,(iii)组织分类(图1)。
首先,图像处理模块将全切片图像(WSI)分割并重新缩放为重叠的图像块,以便进行细胞核定位。细胞核定位阶段使用目标检测模型识别每个图像块中的细胞核。
利用这些细胞核坐标,图像处理模块在每个细胞核周围裁剪图像块,并将其输入到细胞分类阶段。细胞分类阶段使用图像分类模型将细胞核分类为11种胎盘细胞类型之一。
这些细胞被输入到组织分类阶段,成为全切片图像中细胞图的节点,其边被构建为表示同一结构内可能的细胞相互作用。
该细胞图被输入到节点分类图神经网络(GNN),预测每个细胞所属的9种组织显微结构类型之一。
2-1:图像处理
图像处理模块首先根据玻片文件格式选择合适的玻片读取库(libvips⁵⁸、openslide⁵⁹或bioformats⁶⁰)。
将玻片划分为1600×1200像素(177.44×133.08 μm)的图像块,在细胞核定位阶段设置200像素(22.18 μm)的重叠区域,以排除不含组织的区域。
对于细胞分类阶段,以每个细胞核为中心提取200×200像素(22.18×22.18 μm)的图像块。所有图像块均提取并加载到内存中的设备(CPU或GPU)上,无需额外磁盘存储。
考虑到玻片扫描仪的像素尺寸差异,所有图像块均重新缩放为0.1109微米/像素。元数据和结果高效存储于SQLite数据库,并流式传输至HDF5文件,支持全切片图像的推理过程暂停与继续。
2-2:细胞核定位
细胞核定位阶段采用带有200像素(22.18 μm)重叠的1600×1200像素图像块,确保每个细胞核至少完整显示一次,通过后处理去除小半径(4像素)内的重复预测。
使用基于ResNet-101⁸¹主干的RetinaNet⁸⁰模型预测细胞核边界框,以质心坐标作为最终预测。模型首先基于COCO⁸²权重微调40个 epoch,采用Adam⁸³优化器、焦点损失函数,初始学习率0.0001,每20个 epoch衰减0.5。
选择验证集F1分数最高的模型,以0.001学习率全量训练60个 epoch并保存。输入图像进行包括多种H&E特异性染色增强的大量数据扩增(扩增参数细节见补充表6和补充图7)。
在验证集和测试集上,模型性能通过检测质心与手动标注真值点在特定距离(❤️.3 μm)内的F1分数评估,该距离小于典型细胞核半径以处理注释数据中的微小偏差。
使用NVIDIA A100 GPU进行全切片图像推理时,细胞核定位阶段每秒可检测约1000个细胞核。
2-3:细胞分类
细胞分类阶段以每个预测的细胞核为中心,对200×200像素(22.18×22.18 μm)的图像块进行分类,识别11种胎盘细胞类型,包括4种滋养层细胞(合胞体滋养层细胞、细胞滋养层细胞、合胞体节、绒毛外滋养层细胞)、5种绒毛间充质来源细胞(成纤维细胞、霍夫鲍尔细胞、血管内皮细胞、血管肌细胞、未分化间充质细胞)和2种非绒毛细胞(母体蜕膜细胞、白细胞)。
细胞核周围22.18 μm的半径范围足以捕获大多数细胞的完整结构及周围环境信息(如血管内皮细胞附近的红细胞)。模型首先基于ImageNet⁸⁴权重对ResNet-50⁸¹进行60个 epoch微调,采用Adam⁸³优化器、交叉熵损失函数,学习率0.0001,每20个 epoch衰减0.5,选择验证集准确率最高的模型以相同超参数全量训练100个 epoch并保存。
针对类别不平衡问题,训练中对少数类别进行过采样。输入分类模型的图像采用与细胞核检测相同的数据扩增策略(补充表5)。后处理阶段通过k-d树对合胞体节预测进行处理,将孤立节转换为合胞体滋养层细胞,并将50像素半径内邻居少于4个的合胞体节细胞核重新标记为合胞体滋养层细胞,移除4个及以上邻居的簇。
使用NVIDIA A100 GPU进行全切片图像推理时,细胞分类阶段每秒可分类约230个细胞。
2-4:组织分类
组织分类阶段包括细胞图构建和监督图神经网络节点分类,以将每个细胞分类为九种胎盘组织类型之一,包括足月绒毛膜绒毛类型(干绒毛、锚定绒毛、成熟中间绒毛、终末绒毛和绒毛芽)、母胎表面(绒毛膜板和基板/隔膜)以及大量存在时提示病理的区域¹³⁶⁵⁶⁶⁷²(纤维蛋白和无血管绒毛)。
细胞图的节点基于细胞表型阶段的输出定义,节点特征包含来自细胞分类器倒数第二层的64维嵌入向量。连接细胞节点的无向边通过两种边构建算法(k-近邻算法(k=5)⁸⁶和Delaunay三角剖分⁸⁷)的交集并添加自环构建,这种方式既利用了Delaunay三角剖分图的稀疏连接特性,又限制了跨组织边界的边数量,使消息传递模型能够在不同组织微结构内聚合细胞信息,同时考虑组织大小和内部细胞距离的差异。
研究使用随机初始化的归纳式ClusterGCN⁸⁸模型进行训练,该模型包含16个GraphSAGEConv⁸⁹层(每层256个隐藏单元),训练2000个epoch,采用Adam⁸³优化器、自定义加权交叉熵损失函数、0.001学习率、批量大小200(含批量归一化)和400邻居的子图采样大小。
最终保存的模型为在无邻域采样且边构建算法交集取k=8时验证准确率最高的模型。消息传递算法为每个节点采样并聚合最多16条边连接的附近节点特征,模型基于这些聚合特征预测节点的组织类型,通过聚合构成每个组织微结构的细胞社区信息进行决策。使用笔记本电脑CPU进行全切片图像推理时,组织分类阶段每秒可分类约4500个节点。
节点分类方法的关键优势在于允许模型对同一连续结构的不同区域分配不同组织类型。胎盘组织以树状形态生长,因此单个结构的横截面可能包含多种有效分类(如成熟中间绒毛的横截面可能同时存在终末绒毛分支和纤维蛋白,导致三种不同分类)。
此外,绒毛类型的区分并非绝对离散,终末绒毛通过其基质中>50%为毛细血管及存在血管合体膜与成熟中间绒毛区分²⁰²¹³⁸,但成熟中间绒毛的某一节段若毛细血管异常丰富但无血管合体膜,模型将其节点分类为终末绒毛节段虽可能被视为“错误”,但未必不符合生物学实际。
2-5:数据集与数据标注
手动真值训练、验证和测试数据集标注由C.V.使用QuPath⁹⁰完成。为利用相对较少的数据点(<17k)高效训练模型,细胞核定位和细胞分类阶段的数据集通过引导法迭代创建,在新的未见过的玻片图像块中校正先前模型的预测。
训练、验证和测试集按约70/15/15%的比例随机划分(见补充表7),并对各数据集的模型性能进行单独和组合评估。
组织分类阶段的真值标注通过在组织微结构周围绘制粗略边界完成,边界内的细胞核节点被赋予相应组织类别。玻片的验证和测试区域特意选择大于16跳邻域聚合范围且组织微结构分布与训练集相似的区域(见补充图8)。
按区域划分数据集可避免随机节点划分导致的同组织不同细胞信息泄露问题,这与许多图学习数据集随机划分节点的方式不同。
2-6:硬件、软件与库
所有训练和推理均在单块NVIDIA A100 GPU上完成。全切片图像处理使用C库libvips(v8.9.2)⁵⁸,优先调用OpenSlide(v3.4.1)⁵⁹或bioformats(v6.11)⁶⁰,通过其Python绑定pyvips(v2.1.14)、openslide-python(v1.2.0)和python-bioformats(v4.0.7)调用。
代码基于Python(v3.10.13)编写,细胞核定位和细胞表型流水线使用PyTorch(v2.0.1)⁹³和torchvision(v0.15.2)深度学习框架,组织表型流水线采用PyTorch Geometric扩展(v2.3.1)⁹⁴。
结果记录于SQLite数据库(v3.2.7)和HDF5文件(v3.8.0),全切片图像可视化及细胞、组织注释通过QuPath(v0.3.1)⁹⁰完成。
分析和模型训练中使用的其他Python库包括albumentations(v1.3.0)、peewee(v3.16.2)、pytest(v7.3.1)、typer(v0.9.0)、visdom(v0.2.4)、matplotlib(v3.7.1)、pandas(v2.0.1)、numpy(v1.24.1)、scikit-image(v0.22.0)、scikit-learn(v1.3.1)、umap-learn(v0.5.3)和seaborn(v0.12.2)。
2-7:数据与代码可用性
训练和验证各深度学习模型生成的数据集及训练好的模型权重可通过Google Drive链接:https://tinyurl.com/happyplacenta 或Zenodo⁹⁵:10.5281/zenodo.10535021 无限制下载,说明见GitHub README:https://github.com/Nellaker-group/happy。
用于图模型训练的两张组织学玻片可通过CC BY 4.0协议从BioImage Archive(10.6019/S-BIAD1045)下载。其余内部胎盘组织学玻片和临床数据因现有研究伦理委员会批准和数据传输协议限制未公开。
RetinaNet和ResNet-50模型的预训练ImageNet权重通过PyTorch从https://download.pytorch.org/models/resnet101-5d3b4d8f.pth和https://download.pytorch.org/models/resnet50-0676ba61.pth下载,首次使用模型时自动获取。源代码随本文提供。
代码可通过以下GitHub仓库获取:https://github.com/Nellaker-group/happy 及https://doi.org/10.5281/zenodo.1052923996。
三、HAPPY 组织学分析流程解读
HAPPY (Histology Analysis Pipeline.PY) 是一个深度学习驱动的胎盘组织学分析系统,具有三大创新点:
- 生物层次建模:模拟"细胞 → 细胞群落 → 组织结构"的生物层级
- 单细胞分辨率:在全玻片图像(WSI)上实现细胞级分析
- 临床可解释性:提供健康胎盘基线指标,可检测病理偏差(如胎盘梗死)
3-1:环境安装(支持MacOS/Ubuntu)
# 安装libvips图像处理库
# MacOS:
brew install vips --with-openslide# Ubuntu:
sudo apt install libvips# 克隆代码库并创建环境
git clone git@github.com:Nellaker-group/happy.git
cd happy
conda create -y -n happy python=3.10
conda activate happy# 安装依赖(使用CUDA 11.7版本)
make environment_cu117
3-2:数据准备
-
从Google Drive下载胎盘数据集
-
按标准目录结构组织文件:
projects/placenta/ ├── datasets/ │ ├── nuclei/ # 细胞核检测数据 │ └── cell_class/ # 细胞分类数据 ├── annotations/ # 标注文件 │ ├── nuclei/ # 细胞核标注 │ └── graph/ # 组织结构标注 ├── slides/ │ └── sample_wsi.tif # 示例WSI图像 └── embeddings/ # 图模型输入数据
3-3:模型训练
(1) 细胞核检测模型
# 第一阶段:冻结主干网络微调
python nuc_train.py --project-name placenta \--annot-dir annotations/nuclei \--dataset-names hmc uot empty \--frozen --init-from-inc# 第二阶段:解冻网络完整训练
python nuc_train.py --project-name placenta \--pre-trained path/to/阶段1模型 \--no-frozen --no-init-from-inc
(2) 细胞分类模型
# 微调预训练模型
python cell_train.py --project-name placenta --organ-name placenta \--annot-dir annotations/cell_class \--dataset-names hmc uot nuh \--frozen --init-from-inc# 完整训练
python cell_train.py --project-name placenta --organ-name placenta \--pre-trained path/to/微调模型 \--no-frozen
(3) 组织分类模型
python graph_train.py --project-name placenta --organ-name placenta \--run-ids 1 2 \ # 使用两个WSI的数据--annot-tsvs wsi_1.tsv wsi_2.tsv \--val-patch-files val_patches.csv \--test-patch-files test_patches.csv
3-4:全玻片图像分析流程
(1) 添加WSI到数据库
你可以使用 happy/db/add_slides.py 将全切片图像(WSIs)添加到数据库中。这会将指定目录下具有指定文件格式的所有切片添加到数据库中。
根据论文所述,我们在 GitHub 上提供了一个初始数据库,其中的切片表(Slide)和评估运行表(EvalRun)包含两条记录,可用于图模型的训练和评估。
# 获取当前绝对路径
CWD=$(pwd)# 添加WSI到数据库
python happy/db/add_slides.py \--slides-dir "$CWD/projects/placenta/slides/" \--slide-file-format .tif \--pixel-size 0.2277 # 每像素0.2277微米
(2) 运行细胞级分析
python cell_inference.py --project-name placenta \--organ-name placenta \--nuc-model-id 1 \ # 细胞核模型ID--cell-model-id 2 \ # 细胞分类模型ID--slide-id 3 \ # 玻片ID--cell-batch-size 100 # 批处理大小
(3) 运行组织级分析
python graph_inference.py --project-name placenta \--organ-name placenta \--pre-trained-path projects/placenta/trained_models/graph_model.pt \--run-id 3 # 对应cell_inference的运行ID
3-5:结果可视化与导出
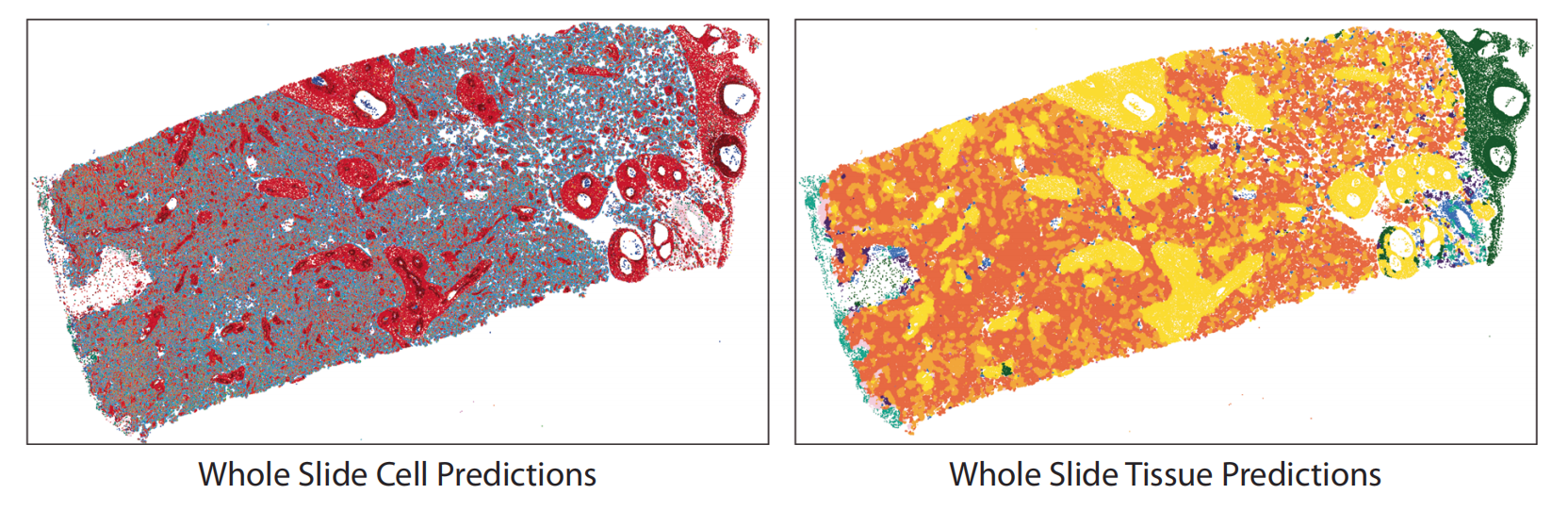
- 细胞级结果:
- 位置:
projects/placenta/predictions/sample_wsi/ - 格式:
.tsv文件(可用QuPath打开) - 包含:细胞坐标、类型、置信度
- 位置:
- 组织级结果:
- 位置:
projects/placenta/trained_models/eval/run_3/ - 包含:
tissue_map.png:组织分区可视化graph_predictions.tsv:组织预测数据cell_graph.gml:细胞图结构文件
- 位置:
四、HAPPY框架解析
HAPPY 实现了一个分层的三阶段深度学习流水线,可对组织学图像进行处理,以识别细胞核、分类细胞并表征组织结构。
尽管设计上具有器官通用性,但当前实现包含针对胎盘组织学分析的特定配置。
该流水线通过在全切片图像上以单细胞分辨率遵循从细胞到细胞群落再到组织结构的生物学层级,解决了组织学分析中组织异质性和变异性的挑战。
4-1:系统架构
HAPPY 的架构遵循分层处理全切片图像的三阶段流水线:
- 细胞核检测:识别全切片图像中的单个细胞核
- 细胞分类:根据形态和外观对检测到的细胞进行分类
- 组织分类:构建表示细胞关系的图以对组织结构进行分类
每个阶段的结果存储在SQLite数据库和HDF5文件中,用于进一步分析和可视化。
4-2:核心组件
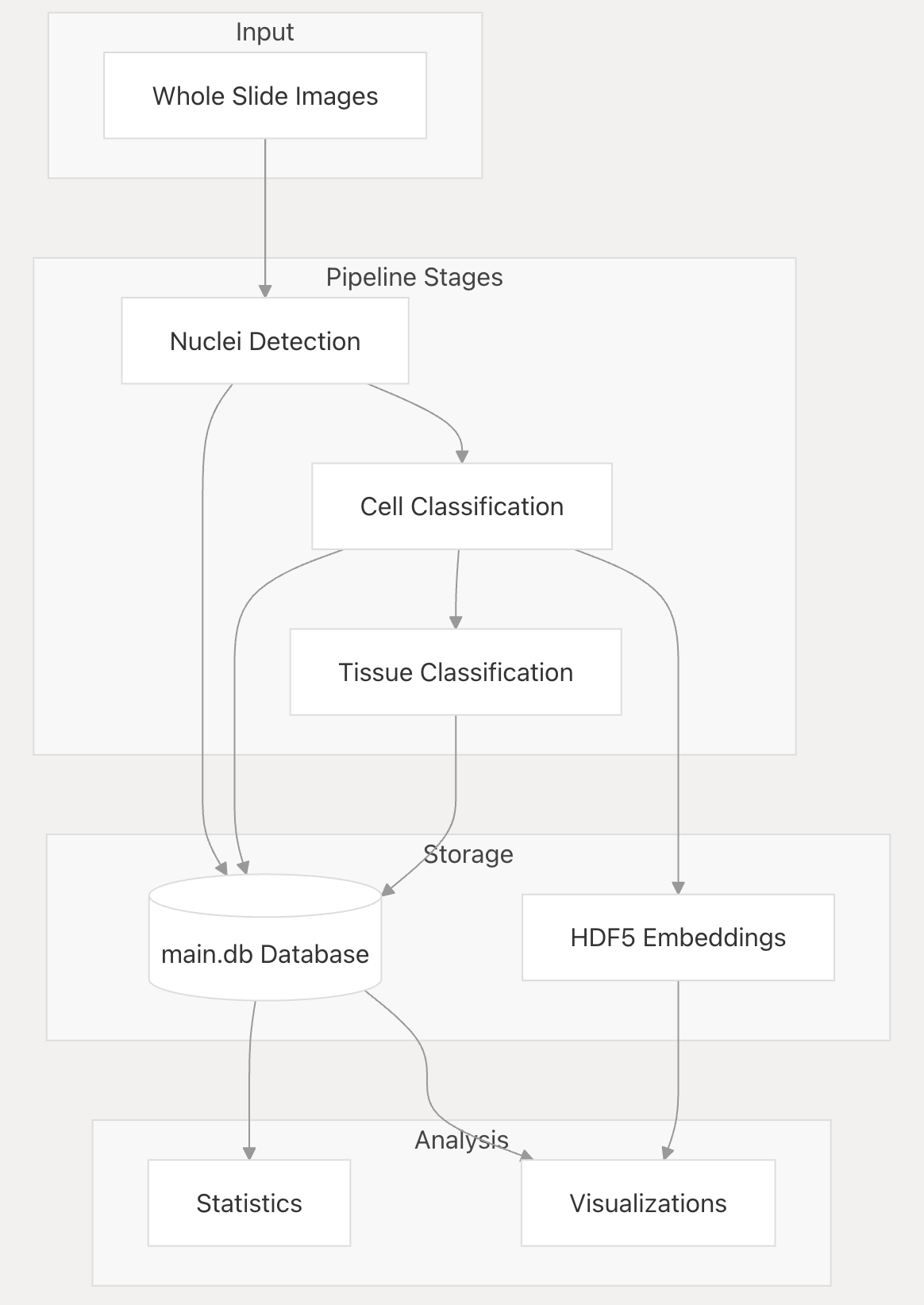
HAPPY 的实现由多个相互关联的组件组成:
WSI处理
利用WSI读取器(OpenSlide、BioFormats、Libvips)读取全切片图像(WSI),通过MicroscopeFile进行相关处理 。
数据管理
- 数据集类(Dataset Classes) :对数据进行分类管理。
- 数据转换(Data Transformations) :对数据进行必要的转换操作。
训练流水线
- cell_train.py:用于细胞分类模型的训练。
- nuc_train.py:用于细胞核检测模型的训练。
- graph_train.py:用于组织分类模型的训练。
训练得到的模型进入推理阶段。
推理流水线
- cell_inference.py:基于训练好的模型,进行细胞核检测和细胞分类的推理,产生细胞嵌入(Cell Embeddings) 。
- graph_inference.py:利用训练好的模型以及细胞嵌入等信息,进行基于图的组织分类推理。
推理结果通过Prediction Saver进行保存。
数据库
推理结果等数据存储到main.db数据库中,数据库通过EvalRun Interface和MicroscopeFile Interface进行相关操作 。
分析
- 图形可视化(Graph Visualization) :对相关数据进行可视化展示。
- 细胞/组织统计(Cell/Tissue Statistics) :进行细胞和组织相关的统计分析。
- 评估脚本(Evaluation Scripts) :运行评估脚本对模型等进行评估。
4-3:数据流与处理
输入处理
- 全切片图像(Whole Slide Image) :作为原始输入数据。
- WSI读取器(WSI Reader) :利用OpenSlide、BioFormats、Libvips等读取器,读取全切片图像。
- MicroscopeFile :对读取的图像数据进行相关处理,生成图像图块(Image Tiles) 。
检测与分类
- 细胞核检测(Nuclei Detection) :使用RetinaNet模型对图像图块进行处理,识别细胞核坐标(Nuclei Coordinates) 。
- 细胞分类(Cell Classification) :基于细胞核坐标,采用ResNet模型对细胞类型进行分类,并生成细胞嵌入(Cell Types & Embeddings) 。
- 图构建(Graph Construction) :根据细胞分类结果构建细胞图(Cell Graph) 。
- 组织分类(Tissue Classification) :运用ClusterGCN模型对细胞图进行处理,完成组织分类 。
存储与分析
- main.db数据库(main.db Database) :存储检测与分类过程中的数据。
- HDF5嵌入(HDF5 Embeddings) :存储相关嵌入数据。
- 分析与可视化(Analysis / Visualization) :基于数据库和HDF5中的数据,进行分析和可视化展示 。
4-4:项目结构
happy/ (Core Library) :核心库文件夹
- db/ (Database) :数据库相关代码,用于存储玻片、模型、评估运行和预测结果等数据 。
- microscopefile/ (WSI Handling) :处理全切片图像(WSI)格式相关的类和代码 。
- models/ (Neural Networks) :定义神经网络模型的代码 。
- utils/ (Utilities) :存放实用函数和辅助类 。
- organs.py (Organ Definitions) :包含器官特定配置的文件 。
projects/ :器官特定项目文件夹,以胎盘(placenta)项目为例
- placenta/ (Example Project) :胎盘项目文件夹
- datasets/ :存放数据集 。
- annotations/ :存储注释数据 。
- slides/ :放置全切片图像玻片相关数据 。
- trained_models/ :保存训练好的模型 。
analysis/ :分析相关文件夹
- evaluation/ :评估脚本存放处 。
- visualization/ :可视化相关代码存放处 。
Pipeline Scripts :流水线脚本文件夹
- nuc_train.py :细胞核检测模型训练脚本 。
- cell_train.py :细胞分类模型训练脚本 。
- graph_train.py :组织分类模型训练脚本 。
- cell_inference.py :细胞核检测和细胞分类推理脚本 。
- graph_inference.py :基于图的组织分类推理脚本 。
该目录结构清晰划分了项目的核心库、特定项目、分析代码以及训练和推理脚本等内容,便于代码管理和项目开发。
结束语
本期推文的内容就到这里啦,如果需要获取医学AI领域的最新发展动态,请关注小罗的推送!如需进一步深入研究,获取相关资料,欢迎加入我的知识星球!